Case Study: Achieving a 505(b)(2) Regulatory Approval for Multiple NDA-Enabling Studies
Hybrid medicines are drugs based on an approved active substance (or previously approved drug) and have a different route of administration, format, strength, or indication from the original reference product.
They require approval for market authorization based on both data from the original reference medicine, and from new clinical trials on the modified version.

In the U.S.A., the 505(b)(2) New Drug Application (NDA) approval process applies to generic molecules with slight modifications from the reference medicine. allows for the approval of drugs that are modifications of previously approved drugs. This process permits the applicant to partially rely on existing data, including published literature, prior FDA findings, and data not developed by the applicant—to support safety and efficacy.
THE PURPOSE OF THE CLINICAL TRIALS
This case study demonstrates how we supported a sponsor pursuing FDA 505(b)(2) approval for a drug intended to treat neurological disorders. To build the regulatory dossier, the required clinical studies were conducted in healthy participants.
The sponsor in this particular study was developing a product that isolated the active enantiomer to make an extended release formulation. These modifications were significant enough to warrant additional clinical studies to help provide a robust dossier for the FDA to review. To support the sponsor, we conducted three key studies:
- A driving simulator study to assess a patient’s ability to drive or operate heavy machinery after an acute dose or at steady state;
- a bioavailability study comparing two lots of extended-release investigational products manufactured via different processes; and
- a study to assess the safety, tolerability, and pharmacokinetics of multiple ascending doses (MAD) of the investigational product from the bioavailability study.
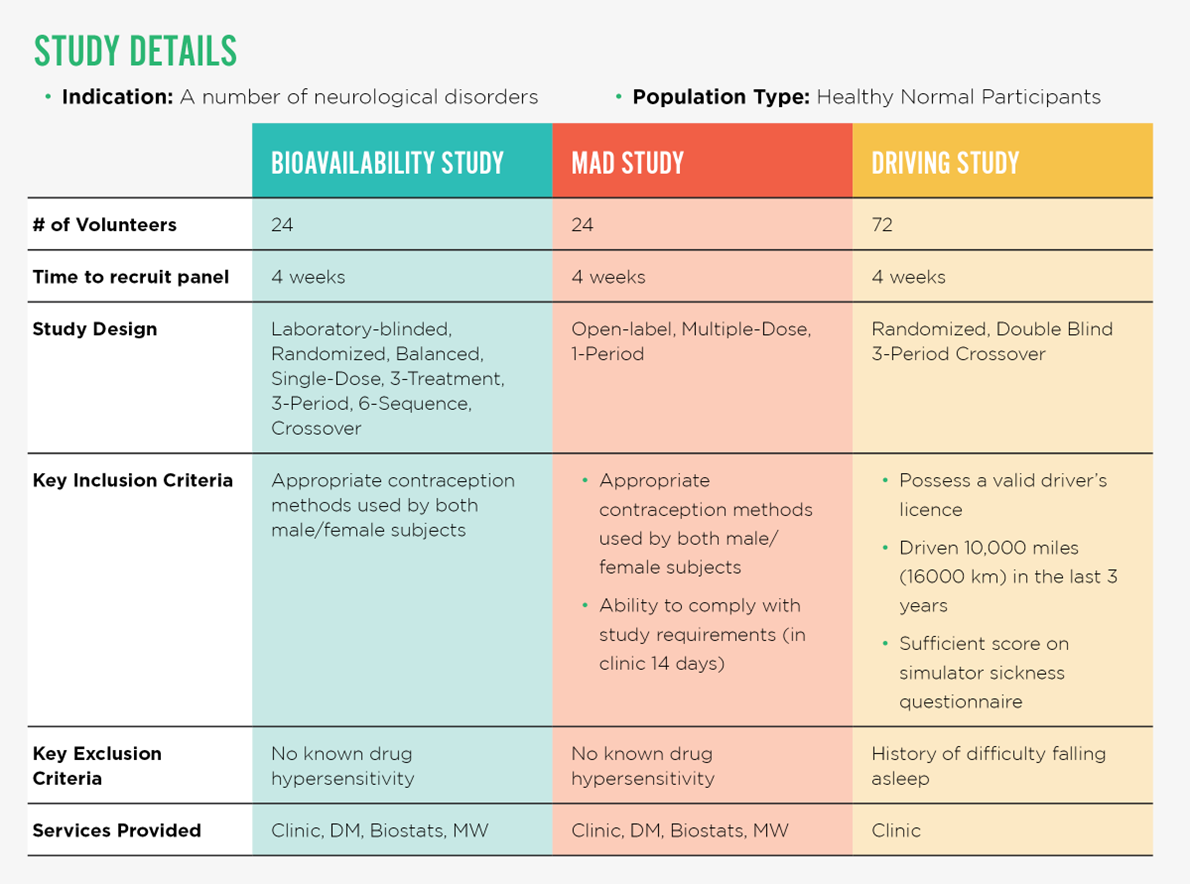
METHODS USED TO SUPPORT THE SPONSOR’S FDA 505(B)(2) APPROVAL
1. Driving Simulation Study
We worked with the sponsor to help design and conduct a driving study that would generate data for a fulsome assessment on the impact of the drug’s sedating effects, and potentially help to justify a modification of the label compared to the reference listed drug (RLD).
The driving study was a randomized, multiple-dose, double-blind, placebo-controlled, Latin-square design with 3-period (full) crossover study, with subjects randomized to treatment sequences (one treatment group per period for three periods). Subjects completed all three treatment periods within the treatment group to which they were randomized.
During each treatment period, participants received either an active treatment (at two dose levels) or a matching placebo twice daily (BID), along with diphenhydramine or a placebo every morning (QAM) for a duration of 15 days. The study drug was administered on-site every four to five days in the morning by site staff on Days 1, 6, 11, 15, 21, 26, 31, 35, 41, 46, 51, and 55. The remaining doses were self-administered by the participants at home on all other study days.
Cognitive testing and driving simulations were conducted approximately 2.5 hours and 3 hours after dosing, respectively. Participants continued their assigned study medication at home, with titration occurring at 5-day intervals.
Before the Day 15 dose for each treatment period, subjects returned to the clinic and stayed overnight. They were dosed the following morning then underwent cognitive and driving simulation testing approximately 2.5 and 3 hours after the morning dose. The evaluations were completed via CogScreen Symbol Digit coding and CRCDS-Mini Sim, respectively.

2. Bioavailability Study
For the relative bioavailability study, both male and female subjects underwent a screening period up to 28 days prior to the study’s conduct to ensure that 24 subjects were enrolled. For all three periods of participant treatment, subjects reported to the clinic on the evening prior to dose administration and fasted overnight for at least 10 hours.
On the morning of the first treatment period, subjects were randomized to either Treatment A, Treatment B, or Treatment C. The study drug was administered following pre-dose clinical assessments and the collection of a blood sample at the 0-hour mark. After dosing, the subjects remained at the clinic for 36 hours, during which blood samples were taken at 14 different intervals. A 7-day washout period was observed between each treatment period.
3. Multiple Ascending Dose (MAD) Study
In the multiple ascending dose study, participants entered the clinic the evening before receiving their first dose. They were given multiple-dose  oral administration of the investigational product, starting on Day 1.
oral administration of the investigational product, starting on Day 1.
Similar to the bioavailability study, participants were admitted to the clinic the night before and were required to fast for at least 10 hours prior to the first drug administration. Before the first dose, they underwent clinical assessments and had a blood draw at 0 hours.
On Day 1, participants received their initial dose of the drug. On Day 4, the dosage was increased by 20 mg, and then again by another 20 mg on Day 7. Dosing occurred in the morning and evening, precisely 12 hours apart.
Pharmacokinetic assessments were conducted throughout the study, during both morning and evening dosing sessions. Participants remained at the study site for the entire duration of the clinical study, which lasted until Day 13. The total length of this study, not including the screening phase, was anticipated to be at least 14 days.
RESULTS
In the driving study, sensitive, objective measures of performance, alertness, and attention confirmed that the initial starting dose was non-sedating. At steady state, treatment with twice the starting dose showed only a mild sedating effect, notably less severe than that caused by a common over-the-counter antihistamine (diphenhydramine HCl 50 mg).
The bioavailability study determined that the rate and extent of absorption were equivalent between the reference product and the extended release manufactured with the alternate manufacturing process.
In the MAD study, systemic exposure of the investigational compound increased proportionally when the dose was doubled. Steady-state disposition was attained within one to two days after initiating twice-daily administration. The study drug was fairly well tolerated at all doses. Increased treatment emergent adverse events (TEAE) were noted at the doubled dose; however, they were consistent with the known safety profile.
ACHIEVING 505(B)(2) APPROVAL FOR OUR CLIENT
We conducted three critical studies to generate the data required for our client’s 505(b)(2) submission: a driving study to assess drug impairment for the product monograph, a bioavailability study comparing two lots of the new formulation, and a multiple ascending dose (MAD) study confirming that dose escalation of the extended-release formulation was safe and well-tolerated.
Our experts worked closely with the sponsor to design protocols, build study schedules, and recruit the right participants. All three studies met their primary endpoints and were delivered on time, providing regulators with essential benefit–risk information. These results highlight our ability to deliver high-quality clinical pharmacology support across diverse study types, aligned with FDA expectations for hybrid medicines.
Key study takeaways:
- a well-planned clinical program can minimize risks and accelerate timelines.
- even small modifications to a known drug may require tailored clinical data.
- close collaboration between sponsor and CRO ensures efficiency and regulatory readiness.
- conducting multiple NDA-enabling studies in parallel can provide a cost-effective path to approval.
Altasciences’ integrated approach and cross-functional expertise were pivotal in generating the data needed to support FDA approval under the 505(b)(2) pathway.
Are you considering a hybrid or 505(b)(2) marketing authorization submission? Contact us with your program needs, and we'll get your required studies up and running, fast.



